
A Sandor Initiative Towards Richer Clinical Insights
CASE BULLETINS
Disclaimer: This newsletter is intended to enrich the insights related to genetic disorders from a laboratory perspective. The objective of this newsletter is to share knowledge from a lab perspective to facilitate the dialogue of genetic disorders diagnosis. We want to sincerely thank the physicians whose dedication, knowledge and intelligence helps arrive at answers through diagnosis enabling timely and effective prevention.
ABOUT THE DISEASE
Mutations in the GAA gene have been found to be associ-
ated with GLYCOGEN STORAGE DISEASE II; GSD2
(OMIM #232300)[1], is an autosomal recessive lysosomal
storage disease. Pompe disease has been classified into:
classic infantile, juvenile and adult forms involvement of
skeletal muscles dominates the clinical picture Matsuishi et
al. (1984).
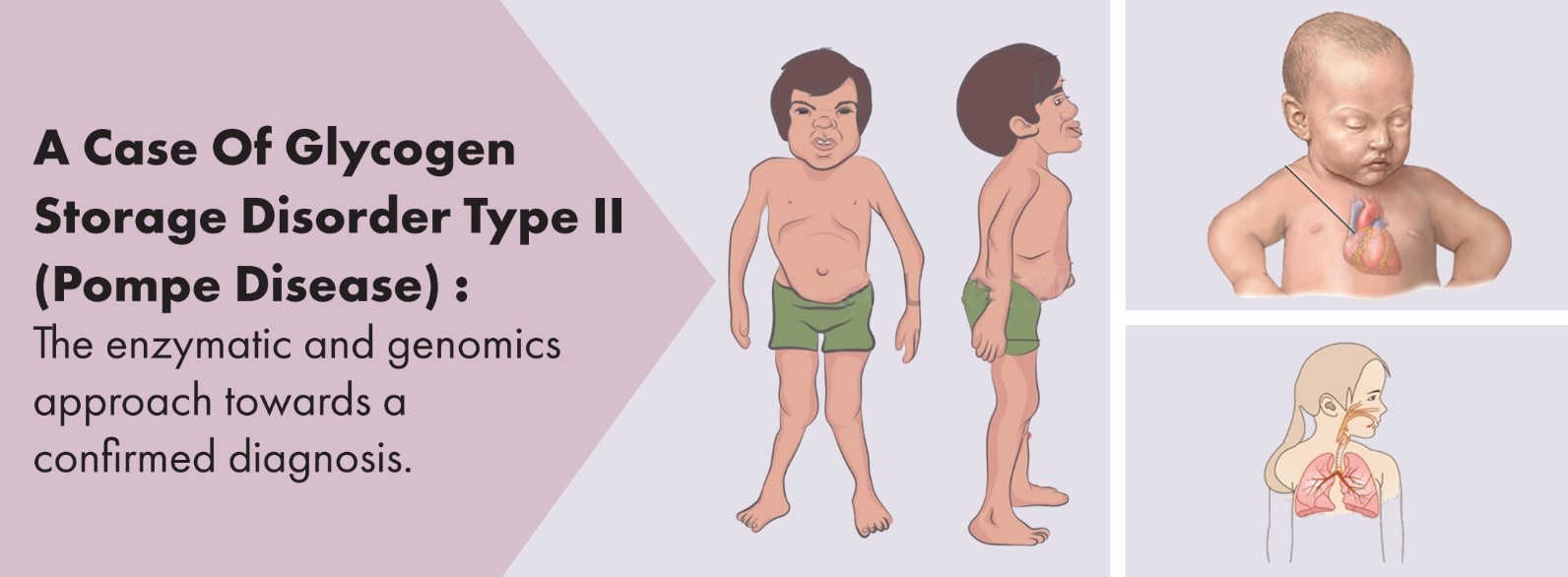
Classic infantile-onset: Age of onset is usually in the first
two to six months of life, with hypotonia and muscle weak-
ness, motor developmental delay, feeding difficulties, fail-
ure to thrive, respiratory distress or infections, cardiac prob-
lems (cardiomegaly, generalised hypertrophy, a murmur
and cardiomyopathy) and hepatomegaly. Children with this
form usually have a severe deficiency of the acid alpha-glu-
cosidase enzyme. If not treated, the symptoms progress
rapidly and the hypertrophic cardiomyopathy may develop
a left ventricular outflow tract obstruction or compress adja-
cent respiratory structures. Cardiorespiratory failure could
result in death within the first year of life.compress adjacent
respiratory structures. Cardiorespiratory failure could result
in death within the first year of life.
INon Classic infantile-onset: Age of onset is within the first
year of life, with motor developmental delay and weakness.
Cardiomegaly is less common, and cardiac involvement is
not present in all the patients. The rate of clinical progression
is slower and without treatment, death will usually occur in
childhood as a result of respiratory insufficiency.
Late or adult onset form: The late-onset type of Pompe
disease may not become apparent until later in childhood,
adolescence or adulthood. Late-onset Pompe disease is
usually milder than infantile-onset and is less likely to in-
volve the heart. Clinical features include proximal muscle
weakness, fatigue, orthopnoea, sleep apnea and respira-
tory failure. Individuals affected by late-onset have a par-
tial deficiency (2-40%) of the enzyme acid alpha-glucosi-
dase. Without treatment, morbidity and mortality occur
mainly as a result of respiratory insufficiency and failure,
with death occurring anytime from the third decade on-
wards mainly as a result of respiratory insufficiency and fail-
ure, with death occurring anytime from the third decade on-
wards.
CASE REFERRED TO SANDOR
Patient born of 3° consanguineous marriage presented with peripheral hypotonia with quadriparesis, hypertrophic car- diomyopathy, and has been evaluated for glycogen storage pompe disease type II.
Tests Prescribed
- Enzyme analysis for Alpha glucosidase
- Clinical Exome Sequencingt
About the Prescribed Tests
| Test Name | Sample Type | Method | Description |
|---|---|---|---|
| Enzyme analysis for Alpha glucosidase | 2-4 ml sodium heparin blood | Fluorometry assay with artificial substrate | Assay carried on leukocytes |
| Clinical Exome Sequencing | 2-4 ml EDTA blood | Next Generation Sequencing | Mean coverage of 80-100X coverage. Target coverage 200X |
Test Results
- Enzyme analysis: Baby was found to have reduced alpha glucosidase enzyme activity (19.1 nmol/hr/mg) vs. biological reference range 56-296 (88.5±23.7) nmol/hr/mg.
- Clinical Exome Sequencing: In Exome data, missense mutations in GAA gene on Exon 19 of Chr 17: 78092588(c.2783A>G / p.Tyr928Cys) and on Exon 6 of Chr 17: 78082136 (c.2783A>G / p.Tyr928Cys) was ob- served and is suspected to be a double or compound heterozygous in Autosomal recessive pattern of inheritance.
Note
In such type of carrier status cases, Validation of the variant by Sanger sequencing and/or Parental segregation analysis can be recommended.
Insights
- The recommended first-tier test for glycogen storage disease type II (Pompe disease) is Alpha glucosidase(acid maltase), Leukocytes .
- Individuals with GAA activity below the reference range for these tests are more likely to have variants in the GAA gene that are identifiable by molecular genetic testing.
- Exome sequencing along with measurement of enzyme activity can significantly improve the diagnostic yield.
This bulletin is compiled by
| Name | Designation | Contribution |
|---|---|---|
| Ms. Bushan Niharika | Research Officer | Compilation |
| Ms. S Ramya | Research Officer | Compilation |
| Ms. Divya Borkar | Sr. Manager, Enzymology | Enzyme Assay |
| Ms. Vineeta Singh | Vice President - Scientific Affairs | Exome Analysis |
References:
- GLYCOGEN STORAGE DISEASE II; GSD2 (OMIM #232300).
- Matsuishi, T., Yoshino, M., Terasawa, K., Nonaka, I. Childhood acid maltase deficiency: a clinical, biochemical, and morphologic study of three patients. Arch. Neurol. 41: 47-52, 1984.[PubMed: 6360103] [Full Text].