
A Sandor Initiative Towards Richer Clinical Insights
CASE BULLETINS
Disclaimer: This newsletter is intended to enrich the insights related to genetic disorders from a laboratory perspective. The objective of this newsletter is to share knowledge from a lab perspective to facilitate the dialogue of genetic disorders diagnosis. We want to sincerely thank the physicians whose dedication, knowledge and intelligence helps arrive at answers through diagnosis enabling timely and effective prevention.

ABOUT THE DISEASE
Phenylketonuria (commonly known as PKU) is an inherited disorder that increases the levels of a substance called phenylalanine in the blood. Phenylalanine is a building block of proteins (an amino acid) that is obtained through the diet. It is found in all proteins and in some artificial sweeteners. If PKU is not treated, phenylalanine can build up to harmful levels in the body, causing intellectual disability and other serious health problems. Mutations in the PAH gene cause phenylketonuria. The PAH gene provides instructions for making an enzyme called phenylalanine hydroxylase. This enzyme converts the amino acid phenylalanine to other important compounds in the body. The most severe form of this disorder is known as classic PKU.
Infants with classic
PKU appear normal until they are a few months old.
Without treatment, these children develop permanent
intellectual disability.Seizures, delayed development, behavioral problems, and
psychiatric disorders are also common. Untreated individuals
may have a musty or mouse-like odor as a side effect of
excess phenylalanine in the body. Children with classic PKU
tend to have lighter skin and hair than unaffected family
members and are also likely to have skin disorders such as
eczema.Less severe forms of this condition, sometimes
called variant PKU and non-PKU hyperphenylalaninemia,
have a smaller risk of brain damage.
People with very mild cases
may not require treatment with
a low-phenylalanine diet.
Tests Prescribed
- New Born Screening/TMS
- Urine Organic Acid Test
- Plasma Amino Acid Test
About the Prescribed Tests
| Test Name | Sample Type | Method | Description |
|---|---|---|---|
| Urine Organic Acid Test | 5-10 ml urine in Sterile container or Urinsoaked on 4 Urine Filter Papers | Gas Chromatography/Mass Spectrometry (GC/MS) | Qualitative report of abnormal levels of organic acids identified |
| Plasma Amino Acid Test | 2-4 ml EDTA Blood | Liquid Chromatography/ Mass Spectrometry (LC/MS | Amino Acid level test in blood |
Test Results
- TMS: TMS of this baby revealed significant(18.05) evaluation of phenylalanine & phenyl: tyrosine ratio.
- Urine Organic Acid Test: Baby was found to have mild to significant elevated levels of phenylalanine metabolite (Phenyllactic acid, Phenylacetic acid, Phenyl pyruvic acid).
- Plasma Amino Acid Test: Plasma amino acidogram shows significant elevation of phenylalanine.
Figure 1: Results Of Urine Organic Test Report
| Compound Name | Citrulline | 41.54 |
|---|---|---|
| Phenylalanine | 1896.41 | 25.9 - 105 |
| Phenylalanine/Tyrosine ratio | 19.07 | 0.29 - 4.31 |
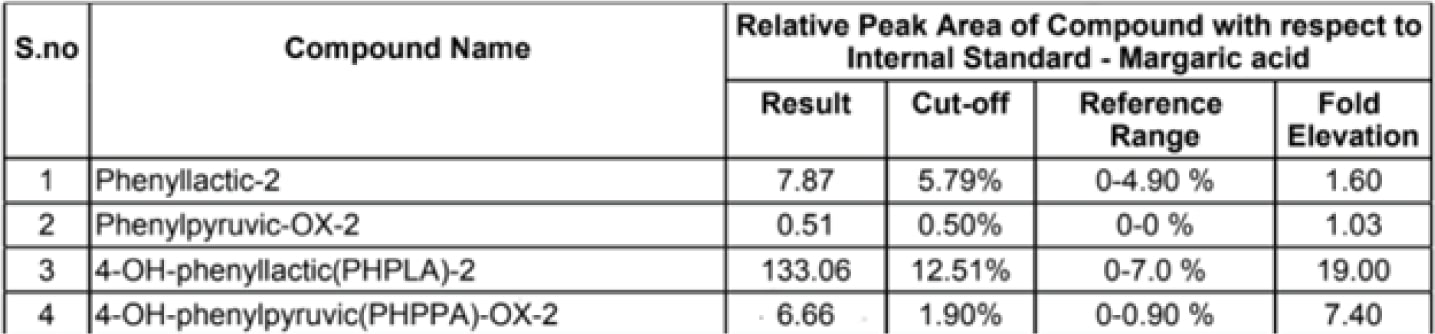
Figure 2: Results Of Plasma Amino Acid Test Report
| 1 | Citrulline | 41.54 | 3-35 |
| 2 | Tyrosine | 46.14 | 22-108 |
| 3 | Phenylalanine | 102.53 | 31-75 |
Insights
- The recommended first-tier test for Phenylketonuria disease is NBS-7, TMS, UOA, followed by PAA.
- Individuals with PKU activity higher than the reference range for these assays are more likely to have variants in the PAH gene that are identifiable by NGS-based Exome Sequencing.
- Urine Organic Acid is the test done to identify any kind of urea cycle disorder.
- Plasma Amino Acid test is done to confirm the levels of phenylalanine and other amino acids to conclude the reports.
- Monitor the treatment by periodically measuring plasma amino acid levels of phenylalanine.
This bulletin is compiled by
| Name | Designation | Contribution |
|---|---|---|
| Ms. Krishnendu Menon | Genetic Counselor | Compilation |
| Mrs. N Sushma Chander | Senior Manager/ QualityCoordinator-Biochemical Genetics | Biochemical Genetics |